Síndrome de
Sinónimos:
Deleción Parcial del Cromosoma 4, 4p
Deleción Parcial del Brazo Corto del Cromosoma 4
Monosomía Parcial del Cromosoma 4, 4p
Wolf, Síndrome deRegión Cromosómica Olf Hirschhorn
4p Parcial,Síndrome
Código CIE-9-MC: 758.3
Descripción en lenguaje coloquial:
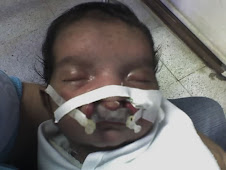
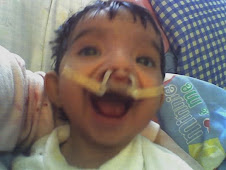
El síndrome de Wolf Hirschhorn es una enfermedad rara del desarrollo, caracterizada por anomalías congénitas múltiples y retraso mental.
Se estima una incidencia de 1 por cada 50.000 recién nacidos.
Clínicamente se caracteriza por una cara peculiar en forma de “casco griego”, microcefalia (cabeza anormalmente pequeña), asimetría craneal, hipertelorismo (aumento de la separación de los ojos), coloboma (fisura congénita en alguna parte del ojo) bilateral, retrognatia (deformidad de la mandíbula, que vista de perfil, parece desplazada hacia atrás), boca en forma de carpa, orejas displásicas (displasia es el desarrollo anómalo de tejidos u órganos) y de implantación baja, convulsiones con inicio temprano generalmente a los 9-10 meses, cardiopatía (término general de la enfermedad del corazón) congénita (que está presente desde el nacimiento): coartación (estrechez) aórtica, comunicación interauricular e interventricular, pene incurvado, hipospadias (apertura urinaria o meatus, que se puede colocar anormalmente en el superficie inferior del pene), hipoplasia (desarrollo incompleto o defectuoso) o ausencia renal en menos del 10% de los casos y retraso mental.
El diagnóstico de sospechaes clínico y el de confirmación se realiza mediante técnicas de citogenética molecular.
El pronostico es grave, alrededor de un tercio de los pacientes mueren antes de los dos años de edad, por las complicaciones broncopulmonares y cardiológicas, se han descrito pocos casos con supervivencia superior a los 10-16 años.
Los supervivientes presentan retraso mental severo y retraso del crecimiento con tendencia a infecciones broncopulmonares de repetición.
Se asocia a una delección en el brazo corto del cromosoma 4 (4p16.3). La mayoría de los casos, 85-90% son delecciones de novo.
Direcciones URL de interés:
Asociaciones:Federación Española de Asociaciones de Enfermedades Raras (FEDER)
Domicilio : c/ Enrique Marco Dorta, 6 local
Localidad: 41018 Sevilla
Teléfono : 954 98 98 92
FAX : 954 98 98 93
European Organization for Rare Disorders (EURORDIS)
Domicilio: Plateforme Maladies Rares 102, Rue Didot
Localidad: 75014 Paris
teléfono : 00 33 1 56 53 53 40
FAX : 00 33 1 56 53 52 15
AVISO IMPORTANTELos datos que se proporcionan UNICAMENTE tienen carácter informativo. En ningún caso la información debe utilizarse con fines diagnósticos ni de tratamiento. No pretende sustituir a la información que le pueda dar su médico. El contenido de esta página puede cambiar sin previo aviso, ya que se pretende mantener la información acerca de estas enfermedades tan actualizada como sea posible.
DELECION 4P
(Sindrome de delecion del brazo corto del cromosoma 4)
Hipertelorismo ocular con nariz ancha o picuda;microcefalia y/o asimetría craneal; oreja simple, de implantacion baja y con hoyuelo preauricular.
ANOMALIAS
Crecimiento: Deficiencia acusada del crecimiento de inicio prenatal, microcefalia.
Funcionalidad: Actividad fetal débil, hipotonía, deficiencia mental grave, convulsiones.
Craneofaciales: Estrabismo, deformidad del iris, hipertelorismo ocular, cejas arqueadas altas, epicanto, glabela prominente, labio leporino y/o paladar hendido, boca en forma de pez y girada hacia abajo, filtro y labio superior cortos, micrognatia, defectos en la linea media del cuero cabelludo posterior, asimetría craneal, hoyuelo o excrecencia preauricular.
Extremidades: Cresta dérmica hipoplasicas, bajo números de crestas dérmicas, pliegues simiescos, pie equinovaro, uñas de las manos hiperconvexas.
Otras: Hipospadia,criptorquidia, fístula u hoyuelo sacro, anomalía cardiaca, defecto del tabique cardiaco (sobre todo entre las auriculas), escoliosis.
ANOMALIAS OCASIONALES
Exoftalmia, ptosis, anomalia de rieger, nistagmo, glaucoma, dientes fusionados, taurodontismo, defecto de la mitad media de las cejas, pérdida de la audición, hipodoncia de los dientes permanentes, linea de implantación de cabello baja con pterigion cervical, metatarso aducto, polidactilia, ectrodactilia, clinodactilia, luxacion de cadera, centro de ocificacion accesoria en los metacarpianos proximales, ausencia de rama púbica, extrofia vesical, retraso de la edad osea, anomalías de los centros de osificacion esternales ,"deformidad en abridor de botellas" de las claviculas, pubertad precoz, anomalía renal, malrotacion del intestino delgado, ausencia del tabique pelucido, quistes interventriculares, sindrome mielodisplasico.
HISTORIA NATURAL
Aunque lo normal es la existencia de retraso mental profundo, en un estudio, el 40% de los pacientes fue capaz de andar, el 20% fue capaz de realizar tareas domesticas sencillas y el 10% pudo controlar sus esfínteres. Las convulsiones inicialmente son dificiles de controlar, pero con la edad tienden a desaparecer. En la infancia, un problema significativo de estos pacientes son las dificultades de alimentarse,por lo que con frecuencia precisan gastrostomía. En esta etapa de la vida, la atención habitual debe incluir evaluaciones cardiacas, oftalmologicas y auditiva, ecografia renal, EEG, estudio de la deglucion y pruebas del desarrollo. Más adelante, estan indicadas las pruebas de desarrollo intelectual, elegir un nivel de educacion adecuado y hacer EEG de seguimiento.
ETIOLOGIA
La causa de este trastorno es una delecion parcial del brazo corto del cromosoma 4. Aproximadamente el 87% de los casos representa deleciones de novo, sin embargo en el 13% uno de los padres es el portador de la transposicion. En los casos que existe la translocacion familiar, se observa un exceso de deleciones 4p de origen materno de dos a uno; en cambio en las deleciones que aparecen de novo el origen del cromosoma es paterno en cerca del 80% de los casos. El fenotipo no difiere segun el tamaño de la delecion,que varía desde casi la mitad del brazo corto del cromosoma hasta un fragmento muy pequeño e indetectable por citogenética. En los casos que existe sospecha clinica del trastorno, pero los estudios cromosomicos estandares son normales, usando un analisis de hibridizacion fluorescente in situ (FISH), con frecuencia se detecta una delecion molecular en el brazo corto del cromosoma 4 a nivel de 4p16.3, la region critica para determinar este fenotipo.
Material encontrado en el libro SMITH Patrones reconocibles de malformaciones humanas, autor kenneth Lyons Jones.
 El Nico ya esta buscando que gorrito ponerse para terminar el año y celebrar el 2009!!!!
El Nico ya esta buscando que gorrito ponerse para terminar el año y celebrar el 2009!!!! 








































































.jpg)

.jpg)


.JPG)